 Previous blog entries have covered what Neurotransmitters are, and how synaptic receptors use these molecules as triggers for complex actions once they have crossed the synapse. This blog entry explores the other side of the synaptic cleft, where the neurotransmitters are stored and released.
Previous blog entries have covered what Neurotransmitters are, and how synaptic receptors use these molecules as triggers for complex actions once they have crossed the synapse. This blog entry explores the other side of the synaptic cleft, where the neurotransmitters are stored and released. Nature has developed some amazing machinery to make synapses fire quickly. Relying on the cell nucleus to generate the molecules that act as neurotransmitters when they are needed would be far too slow to achieve the kind of speed required to power thought. Instead, these molecules are made ahead of time, before they are needed, and are kept bottled up in spherical containers called 'vesicles', waiting for the instant that they need to be released. The picture that is evolving on how synapses actually manage this feat is quite fascinating. And, once again, that super cool molecule called 'clathrin' plays a major role...
From http://www.hhmi.org/research/investigators/sudhof.html:
Neurons communicate with each other by synaptic transmission at specialized intercellular junctions called synapses. During synaptic transmission, a presynaptic neuron releases a chemical neurotransmitter that is then recognized by the postsynaptic neuron.
The electrical impulses, called action potentials, cause the vesicles to fuse with the nerve cell membrane and spill their contents into the synapse. The neurotransmitters then activate receptors on the other side of the synapse, essentially allowing the action potential to “jump the gap” to the next nerve cell.
Neurotransmitter release is triggered when an electrical impulse that travels down the neuron's axon, called an action potential opens voltage-gated Ca2+ (calcium ion) channels and Ca2+ flows into the presynaptic terminal. Ca2+ triggers neurotransmitter release by stimulating the fusion of synaptic vesicles ( [tiny spheres] that are filled with neurotransmitters) with the presynaptic plasma membrane at the active zone (a specialized plasma membrane section that marks the synapse). Released neurotransmitters then elicit a postsynaptic signal. Synaptic transmission occurs by this mechanism in all synapses, but the specific properties of synaptic transmission vary among synapses.
From the NIH:Likened to soap bubbles merging, or bubbles bursting at the surface of boiling water, membrane fusion has attracted heightened interest among neuroscientists in recent years. The family of proteins involved, SNARES (Soluble NSF Attachment protein REceptor), have been conserved through evolution, performing similar functions even in primitive organisms like yeast and fruit flies. Some of the fusion proteins are embedded in the vesicle and the cell wall membranes; others float freely within the nerve terminal, or axon.
The entire neurotransmission performance — from electrical signal to receptor binding — lasts less than a thousandth of a second, and is repeated billions of times daily in each of the human brain's 100 billion neurons. So membrane fusion mechanisms may hold clues about what goes wrong in disorders of thinking, learning and memory, including schizophrenia and other mental illnesses thought to involve disturbances in neuronal communication.
The Synaptic Vesicle In Action!
From http://origins.swau.edu/papers/complexity/trilo/gifs/vesicles.html:
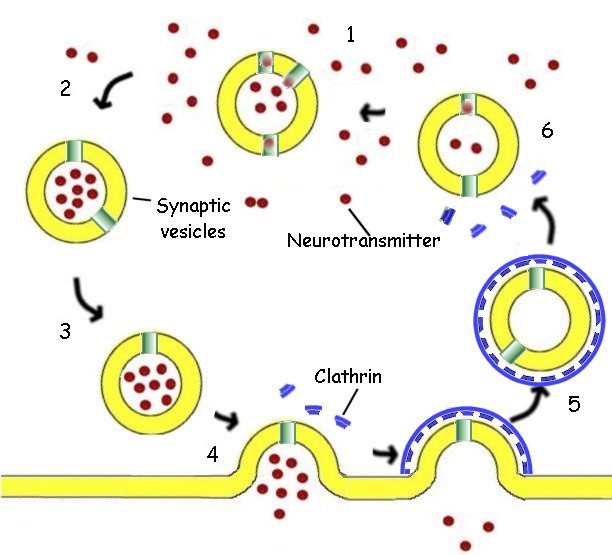
1. The synaptic vesicle accumulates hydrogen ions at the expense of ATP. The hydrogen ions are then exchanged for a neurotransmitter (red) such as acetylcholine, by a specific antiport protein.
2. As the vesicle becomes charged with the neurotransmitter, it is picked up by a cytoplasmic transport protein and carried through the cytoplasm to the synaptic region of the neuron.
3. The filled vesicle is docked in the region of the synapse, where it awaits a nerve stimulation.
4. Triggered by an influx of calcium ions, the vesicle releases its charge of neurotransmitter to the synapse, passing the impulse on to the post-synaptic cell.
5. The empty vesicular membrane is surrounded by cytoplasmic protein molecules called clathrin (blue), capturing the vesicle for reuse. The clathrin forms a cage coating the entire vesicle. The vesicle then travels back away from the membrane into the cytoplasm and loses its protective cage.
6. The vesicle again begins to accumulate hydrogen ions the cycle is repeated.
Nothing is left to chance in this sequence. Each step governed by specialized molecules.
From http://www.cnsforum.com/imagebank/item/vesicle_fusion/default.aspx :
Fusion of a synaptic vesicle with the pre-synaptic membrane

Neurons communicate with their target cells primarily through the regulated fusion of synaptic vesicles with the nerve terminal membrane and subsequent release of chemical neurotransmitter into the synaptic cleft. Synaptic vesicles move down the axon and bind to release sites on the pre-synaptic membrane via vesicle-membrane proteins (v-SNARE) and target-membrane proteins (t-SNAREs). This SNARE complex interacts with both NSF (N-ethylmaleimide Sensitive Fusion protein) and SNAP (Soluble NSF Attachment Proteins) to form a fusion complex. Action potential propagation induces calcium influx at the pre-synaptic membrane, which, in addition to ATP hydrolysis by NSF, results in disassembly of the SNARE complex and membrane fusion. Following neurotransmitter release, synaptic vesicle membrane components are recycled via an endocytic process.
More from http://www.hhmi.org/research/investigators/sudhof.html
Model for the functions of SNARE proteins, complexins, and synaptotagmins 1 and 2 in synaptic vesicle exocytosis.

In docked vesicles (left), SNAREs and synaptotagmins are not engaged in direct interactions. During priming (center), SNARE complexes form, and synaptotagmins constitutively associate with the assembled SNARE complexes. The approximation of the synaptic vesicle and plasma membranes forced by SNARE complex assembly is proposed to create an unstable intermediate that is shown as a fusion stalk, but could have other structures. Subsequently, complexins (green) are bound to fully assembled complexes (right), and calcium influx (right) destabilizes the fusion intermediate by triggering the C2-domains of synaptotagmins to associate with, and partially insert into, the phospholipids. This is proposed to cause a mechanical perturbation that opens the fusion pore. Note that SNARE complex assembly in priming (center) is suggested to be reversible, whereas calcium triggering is not.

At a synapse, pre- and postsynaptic compartments are linked by trans-synaptic cell adhesion molecules that in turn are coupled to the presynaptic release machinery or to postsynaptic receptors. A decade ago, we identified candidate trans-synaptic cell adhesion molecules called neurexins and neuroligins. Neurexins are thought to be presynaptic, are expressed in two principal forms, a- and ß-neurexins, and are highly polymorphic due to extensive alternative splicing. Neuroligins are primarily postsynaptic and are also alternatively spliced; their importance is highlighted by the finding that neuroligin mutations are pathogenic in a subset of patients with familial autistic syndrome.
Neurexins bind to neuroligins to form heterophilic intercellular junctions at the
synapse; this binding is tightly regulated by alternative splicing of neurexins and—as we recently observed—of neuroligins, resulting in a trans-synaptic splice code. Moreover, recent experiments with mutant mice revealed that neurexins and neuroligins are not required for the establishment of initial synaptic contacts, but are both essential for the regular function of synapses. Mice with mutant neurexin or neuroligin genes form ultrastructurally normal synapses. In the mutants, however, synaptic transmission is severely impaired, such that the mutant mice die at birth. Overall, these studies showed that neurexins and neuroligins are genuine synaptic cell adhesion molecules that guide synapse formation not by triggering the assembly of synaptic junctions but by recruiting crucial components of the pre- and postsynaptic machinery.
Neurotransmitter Release
To identify key molecules involved in release, we initially set out to characterize the proteins of synaptic vesicles and of the active zone. We then used biochemical and biophysical methods to determine the properties and atomic structures of these proteins, and targeted mouse mutants to examine their functions. Although many questions remain, this combination of approaches has elucidated fundamental mechanisms underlying key aspects of neurotransmitter release (e.g., Ca2+ triggering of fast synaptic vesicle fusion).
Synaptic vesicle fusion is at least in part mediated by the assembly of three synaptic SNARE proteins—the vesicle protein synaptobrevin/VAMP and the plasma membrane proteins SNAP-25 and syntaxin/HPC-1—into a tight complex. Syntaxin also interacts with another essential fusion protein, Munc18-1. SNARE proteins and Munc18-1 perform multiple functions that are exquisitely regulated. For example, during fusion, syntaxin changes from a closed into an open conformation. We recently showed in mice that permanent "opening" of syntaxin by mutagenesis destabilizes synapses, leading to massive epilepsy. As another example, synaptobrevin is required for both spontaneous and evoked synaptic vesicle fusion, but recent data reveal that the mechanisms by which synaptobrevin acts in these two forms of fusion differ. In addition to this dual function in exocytosis, synaptobrevin is also required for normal fast endocytosis of vesicles. These findings suggest an unexpectedly economical organization of the secretory machinery in which different steps are mediated by the same molecules via different mechanisms.
At the synapse, Ca2+ triggers both a fast component and a slow component of release. The fast component is induced by Ca2+ binding to the synaptic vesicle proteins synaptotagmin 1 and 2. Recent data show that these two synaptotagmins act similarly in stabilizing resting synapses and in triggering fast release upon Ca2+ binding.
...
Active-zone proteins not only serve as a receptacle of synaptic vesicles for fusion but also regulate synaptic vesicle fusion during plasticity. Key active-zone proteins for both functions are Munc13s and RIMs, multidomain proteins that bind to each other and to other active-zone and synaptic vesicle proteins. Many isoforms of Munc13s and RIMs with differential properties are expressed, but all isoforms appear to function in release. Recent results, for example, demonstrated that deleting one RIM isoform, RIM1a, leads to discrete changes in both vesicle priming and the plasticity of release, whereas deletion of multiple isoforms almost completely abolishes release. These and other findings have led to the view of the active zone as a protein mosaic whose precise composition is a key determinant of the properties (for example, strength and plasticity) of a synapse.
...
Several facts — for example, the critical role of the presynaptic protein a-synuclein in the pathogenesis of Parkinson's disease, or the transport of the cell-surface protein APP (the amyloid-ß precursor protein that is critical for the pathogenesis of Alzheimer's disease) into presynaptic nerve terminals — point to a presynaptic dysfunction in at least some neurodegenerative disorders. To approach this problem, we have begun to study the normal synaptic function of APP and synuclein. Our studies have uncovered an unexpected role for APP in gene expression, and led to a description of a powerful activity of synucleins in suppressing neurodegeneration. Strikingly, we found that synucleins genetically interact with CSPa (cysteine string protein a), a synaptic vesicle cochaperone protein. CSPa acts as a proprietary chaperone on synaptic vesicles whose deletion causes massive neurodegeneration. Moderate overexpression of synucleins abolishes the neurodegeneration in CSPa-deficient mice, while deletion of endogenous synucleins accelerates the neurodegeneration.
From the NIH: Molecular Minuet of Membrane Fusion Proteins Unveiled
Just before a neuron sprays a neurotransmitter into the synapse, mercurial molecules perform a secret dance that fuses the vesicle membrane with the cell wall membrane. Proteins change their shapes and locales, as their Velcro-like extensions intertwine to conjure a momentary dragon. This highly charged core complex melds the membranes, then vanishes. In the March 23, 2000 issue of Nature, NIMH grantees Drs. Richard Scheller, William Weis and colleagues at Stanford University reveal, for the first time, the atom-by-atom, 3-D structure and cagey choreography of a lead hoofer in this act, the Sec1-syntaxin1a complex.
...
Scheller, Weis and doctoral student Kira Misura were especially interested in a complex created when two proteins bind together: the free-floating SNARE regulator nsec1, and the target membrane's sharply-bent, helical SNARE, syntaxin1. Formation of the complex is the first act in the membrane fusion ballet. To reveal the complex's secret moves, the researchers examined its crystalline structure using a type of subatomic particle accelerator called a synchrotron. The mile-long accelerator generates very intense x-ray beams used to resolve objects as small as atoms, and capture snapshots of the shapes of molecules at different stages of chemical reactions.
Based on the revealed physical properties, as well as genetic and biochemical evidence, the investigators propose that sec1/syntaxin1 complex communicates with Rab, a G protein on the vesicle membrane, and Rab-E, a free-floating cousin, to recognize and guide the neurotransmitter-filled sac to its appointed fusion site on the target membrane. They further propose that syntaxin1a and Sec1 collaborate with VAMP, a velcro-like vesicle membrane protein, and SNAP-25, a counterpart on the cell's membrane, to bring about fusion itself.
In this model, nSec1 performs a "chaperone-like" role for syntaxin1a, first shielding it from other proteins, and later facilitating its interactions with them. When in its protective posture, nSec1 renders its partner physically incapable of forming stable relationships with other proteins.
The steps to the dance are as follows (See diagram of model below):

A – nSec1 binds tightly to, and cloaks, syntaxin1a, forming a complex that serves as a recognition site for an arriving vesicle.
B – The vesicle's Rab and/or Rab-E proteins recognize the complex, signaling nSec1 to change its shape and move outward, uncovering a key binding region of syntaxin1a, and destabilizing it. Residues from the suddenly exposed binding region signal SNAP-25 to begin making the core fusion complex, using nSec1 as a platform.
C – Syntaxin1a's sharply bent end section moves away from its key binding region, making way for SNAP-25 and VAMP to move in and bind.
D – VAMP/SNAP-25/syntaxin1a combine to form a long, straight, helical bundle, the core complex. This pulls the vesicle and target membranes together, fusing them, and releasing the neurotransmitter. After fusion, an enzyme breaks down the complex and the SNAREs are recycled for a repeat performance.
Discovery sheds light on neurotransmitter release mechanism
Researchers at three medical centers including Vanderbilt have discovered an important mechanism controlling the release of neurotransmitters and hormones. The discovery, reported in the April and May issues of the journal Nature Neuroscience, could lead to new ways to treat pain, Parkinson's disease and perhaps even diabetes.
...
Nature modulates the supply of neurotransmitter through G protein-coupled receptors. These receptors are named for their ability to activate G proteins — actually, to split them into two active parts, an alpha subunit and a beta-gamma subunit.
Four years ago, Hamm and colleagues including Simon Alford, Ph.D., of the University of Illinois at Chicago, and Thomas F. J. Martin, Ph.D., of the University of Wisconsin-Madison, reported that the beta-gamma subunit of an inhibitory G protein blocked neurotransmitter release from large, easily studied nerve cells of the lamprey eel. But they didn't know the mechanism — until now.
In an elegant series of experiments, the researchers showed that the beta-gamma subunit prevents vesicles from fusing with the membrane of the nerve cell. It does this by preventing another protein from binding to the cell's “fusion machinery,” a complex of proteins called SNARE that links the vesicle to the membrane. In particular, the beta-gamma subunit prevents synaptotagmin, a vesicle protein, from binding to the SNAP-25 protein in the SNARE complex.
The researchers identified the site of the subunit's action by injecting tiny amounts of a form of the paralyzing botulinum toxin that chopped off the end of the SNAP-25 protein. When that happened, the subunit could not block fusion, indicating that the chopped-off portion was its binding site.
...
Inhibiting vesicle fusion may be a major way the body regulates the release of a whole array of chemical messengers.

At left, activation of a G protein-coupled receptor releases the beta gamma subunit of an inhibitory G protein. The subunit blocks a vesicle protein, synaptotagmin, from binding to SNAP-25, part of the SNARE complex of proteins (right). This, in turn, prevents the complex from pulling the vesicle and the membrane together. The vesicle is thus unable to fuse to the membrane or spill its contents – neurotransmitter – into the synapse.
by Bill Snyder (May 13, 2005) http://www.mc.vanderbuilt.edu
Comments