 Recall from the previous post, that when a neuron's axon fires repeatedly the relevant genes (in that neuron) turn on, and the synapses that are holding the short-term memory when the synapse strengthening proteins find them, become, in effect, tattooed (from Making Memories Stick by R. Douglas Fields)
Recall from the previous post, that when a neuron's axon fires repeatedly the relevant genes (in that neuron) turn on, and the synapses that are holding the short-term memory when the synapse strengthening proteins find them, become, in effect, tattooed (from Making Memories Stick by R. Douglas Fields)It appears that this 'tattooing' process involves enzymes that cause actin to change the shape of the synapse, broadening it so that more receptors can be brought into play. Much progress has been made in the past 10 years or so to understand the details of what is going on.
From ScienceDaily (Jun. 14, 2004): Neuroscientists at the Picower Center for Learning and Memory at MIT show for the first time that storage of long-term memories depends on the size and shape of synapses among neurons in the outer part of the brain, the cerebral cortex.
...
When an experience or a fact is repeated enough or elicits a powerful emotional response, it shifts from short- to long-term memory. It moves from the hippocampus, in the innermost fold of the temporal lobe, to the brain’s outermost region, the cortex, which controls higher functions like abstract thought and speech. MIT researchers studied how structural and functional alterations of synapses--physical and chemical connections among neurons--in the cortex affect the animal’s ability to store long-term memory.
The MIT research provides the first evidence that synaptic structure and function in the cortex are critical for their long-term storage as memories are transferred from the hippocampus to the cortex.
PAK (p21-activated kinase), a critical regulator of synaptic architecture, was inhibited in the mutant mice. In humans, the gene that encodes this enzyme is tied to mental retardation. "Overall, we believe that one of the underlying mechanisms for mental retardation is synaptic malformation that leads to cognitive dysfunction," Hayashi said. "One of the most significant findings of this study is that the structure of synapses links to the function of synapses, and the size of synapses reflect the strength of the synapse. The bigger the synapse, the more vesicles to carry more neurotransmitters and the more channel proteins to permeate and bind to the neurotransmitters."
From Science Daily: University of California, Irvine researchers, using newly developing microscope techniques, have captured first-time images of the changes in brain cell connections following a common form of learning.
Detailed in the Journal of Neuroscience, the study shows that synaptic connections in a region of rats’ brains critical to learning change shape when the rodents learn to navigate a new, complex environment. In turn, when drugs are administered that block these changes, the rats don’t learn, confirming the essential role the shape change plays in the production of stable memory. “This is the first time anyone has seen the physical substrate, the ‘face,’ of newly encoded memory." said Gary Lynch, professor of psychiatry and human behavior at UC Irvine and leader of one of the two research teams involved in the studies.
The new behavioral experiments followed a series of recent discoveries by the UC Irvine group concerning the synaptic changes responsible for long-term potentiation (LTP), a physiological effect closely related to memory storage. Those studies used brain slices from rats, maintained alive in experimental chambers, to identify chemical markers for synapses that had recently experienced the LTP effect. The new study used live animals.
Working with advanced microscopic techniques called restorative deconvolution microscopy, the UC Irvine team found that the LTP-related markers appear during learning and are associated with expanded synapses in the hippocampus. Because the size of a synapse relates to its effectiveness in transmitting messages between neurons, the new results indicate that learning improves communication between particular groups of brain cells.
Because proteins continually biodegrade, ongoing maintenance operations need to be performed within the synapse or else it will not keep its shape. Prof. Yadin Dudai and his colleagues at the Neurobiology Department of Weizman Institute of Science have found that the process of storing long-term memories involves a miniature molecular machine that must run constantly to keep memories going. They also found that jamming the machine briefly can erase long-term memories. Their findings, which appeared today in the journal Science, may pave the way to future treatments for memory problems.
Dudai and research student Reut Shema, together with Todd Sacktor of the SUNY Downstate Medical Center, trained rats to avoid certain tastes. They then injected a drug to block a specific protein into the taste cortex – an area of the brain associated with taste memory. They hypothesized, on the basis of earlier research by Sacktor, that this protein, an enzyme called PKMzeta, acts as a miniature memory 'machine' that keeps memory up and running. An enzyme causes structural and functional changes in other proteins: PKMzeta, located in the synapses – the functional contact points between nerve cells – changes some facets of the structure of synaptic contacts. It must be persistently active, however, to maintain this change, which is brought about by learning. Silencing PKMzeta, reasoned the scientists, should reverse the change in the synapse. And this is exactly what happened: Regardless of the taste the rats were trained to avoid, they forget their learned aversion after a single application of the drug. The technique worked as successfully a month after the memories were formed (in terms of life span, more or less analogous to years in humans) and all signs so far indicate that the affected unpleasant memories of the taste had indeed disappeared. This is the first time that memories in the brain were shown to be capable of erasure so long after their formation.
'This drug is a molecular version of jamming the operation of the machine,' says Dudai. 'When the machine stops, the memories stop as well.' In other words, long-term memory is not a one-time inscription on the nerve network, but an ongoing process which the brain must continuously fuel and maintain. These findings raise the possibility of developing future, drug-based approaches for boosting and stabilizing memory.
Oster, Eichele and Leitges of The Max-Planck Institue provide more info on PKMzeta here. They've found that PKMzeta is found to be broadly expressed in most of the cortex, the limbic system, and the thalamus.
Another key protein is a-CaMKII. From ScienceDaily (May 21, 2001) A study by scientists from UCLA and Johns Hopkins University reveals the role of a protein that must be present in the cortex for information to be converted from short-term into lifelong memories. It indicates that the a-CaMKII protein triggers changes in cell-to-cell communication needed for establishing permanent memories in the cortex.
This was followed up by research from Yale - ScienceDaily (Mar. 16, 2005): A family of proteins that help build the cytoskeleton, or the bones of the cell, also play an important role in learning and memory, according to a study published in the Journal of Neuroscience 25: 2138–2145 (February–2005)
“If you learn to do something new, your neurons have to adapt and change to create a stronger, more direct pathway between neurons,” Picciotto said. “The protein ß–adducin appears to be important for making those new connections.”
In this study, the mice that did not have the protein were not able to strengthen a synapse in the hippocampus, which is the area of the brain that enables us to remember people, places and things. “If the mice don’t have ß–adducin, they can’t make a new map,” Picciotto said. “It’s not enough to just have the electrical properties, the (cyto)skeleton is very important in making long–lasting changes between nerve cells that result in learning.”
Nature has a review called "Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy" by Cingolani and Goda that ties together many of these discoveries into a coherent picture: Synapse regulation exploits the capacity of actin to function as a stable structural component or as a dynamic filament. Beyond its well-appreciated role in eliciting visible morphological changes at the synapse, the emerging picture points to an active contribution of actin to the modulation of the efficacy of pre- and postsynaptic terminals. Moreover, by engaging distinct pools of actin and divergent signalling pathways, actin-dependent morphological plasticity could be uncoupled from modulation of synaptic strength.
From the corrected version of Box 3 of the article: Synaptic activity regulates actin dynamics and spine morphology through multiple signalling pathways. Key regulators of actin polymerization are the GTPases of the Rho family and serine/threonine kinases, which ultimately target actin-binding proteins (Table 1).
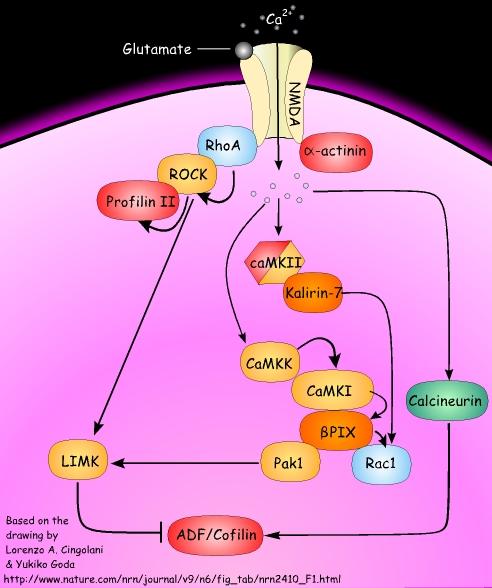
In the figure, actin-binding proteins are shown in red, Rho GTPases are shown in blue, kinases are shown in golden yellow, phosphatases are shown in green and GEFs are shown in dark orange.
In dendritic spines, opening of the Ca2+-permeable NMDA (N-methyl-D-aspartate) receptors by repetitive synaptic stimulation leads to the elevation of intracellular Ca2+ and the subsequent activation and translocation of Ca2+/calmodulin-dependent protein kinase II (CaMKII) to the postsynaptic density. Once activated, CaMKII can phosphorylate and activate multiple downstream signalling targets.
In addition, CaMKII, which is present in dendritic spines at high levels, has been proposed to also function as a structural molecule that can bind to and bundle F-actin through its subunit. Among the intracellular signalling events that are relevant for actin dynamics, of particular interest is the activation of the calmodulin-dependent kinase kinase (CaMKK)–CaMKI signalling pathway. These kinases form a multimolecular complex with the guanine-nucleotide exchange factor (GEF) PIX.
CaMKI-mediated phosphorylation of PIX increases its GEF activity, resulting in the activation of the small GTPase Rac1. PIX also interacts with p21-activated kinase 1 (Pak1), which can be activated by Rac1-triggered autophosphorylation.
In turn, Pak1 phosphorylates several downstream signalling molecules that directly modulate F-actin. For instance, Pak1-mediated phosphorylation activates LIM kinase (LIMK). The only known targets of LIMK are the members of the ADF/cofilin family (actin-depolymerizing proteins), which are inactivated upon LIMK-mediated phosphorylation.
Alternatively, Rac1 can be activated by a second GEF, kalirin 7, which interacts with and is phosphorylated by CaMKII. The activation of LIMK can also be promoted by the small GTPase RhoA, through RhoA-specific kinase (ROCK).
Interestingly, RhoA interacts with NMDA receptors, ROCK and the actin-polymerizing protein profilin II in an activity-dependent manner. Also,alpha-actinin, which is involved in crosslinking and bundling actin filaments, associates with NMDA receptors in an activity-dependent manner. When NMDA receptors open in response to stimuli delivered at low frequency to induce long-term depression, the effects on actin dynamics are opposite to those of CaMKII-initiated pathways, as ADF/cofilin family proteins are turned on following the activation of the phosphatase calcineurin.
More...
ScienceDaily (Nov. 10, 2000): info on NMDA receptors
Comments